Medical Device Prototyping by Class I, II & III: Timeline & Cost in 2026
Stop rebuilding medical device prototypes mid-cycle. Learn how device class determines your timeline, testing, materials & DHF requirements. FDA-aligned.
Stop rebuilding medical device prototypes mid-cycle. Learn how device class determines your timeline, testing, materials & DHF requirements. FDA-aligned.

Medical device prototyping is the structured process of building and testing physical representations of a medical product before full-scale manufacturing. Your device's FDA classification Class I, II, or III determines how you prototype, what materials you use, what tests you run, what you document, and how many iterations you need.
Get the classification wrong early, and you don't just face regulatory delays. You often rebuild from zero.
Why? Here's what most medtech founders discover too late:
A startup builds a connected health monitoring wearable. They prototype it fast, iterate three times, get the hardware working, and then hand everything to a regulatory consultant. Six weeks later, they find out their device is a Class II product requiring 510(k) clearance and that none of their prototype documentation is in a format the FDA will accept. They are now rebuilding their design history from scratch, twelve months behind schedule.
This is not a compliance failure. It is a prototyping strategy failure.
Medical device prototyping is the structured process of building and testing physical representations of a medical product before full-scale manufacturing. What that process looks like - the materials, the tests, the documentation - is dictated entirely by whether your device is Class I, II, or III. Get the classification wrong early, and you do not just face regulatory delays. You often rebuild from zero.
Here is what every founder, R&D manager, and product lead needs to understand before they build product prototype #1.
✓ Your device's FDA class must be determined before you begin prototyping it defines your materials, testing, and documentation requirements from day one.
✓ Class I devices allow fast iteration with representative materials; Class II requires near-final materials at the validation stage; Class III demands production-equivalent materials for any prototype used in preclinical testing.
✓ The Design History File (DHF) is a live document built during prototyping not a pre-submission cleanup task.
✓ For Class III, the prototype is effectively the regulatory artifact. Every engineering decision must be defensible in a PMA submission.
✓ ISO 13485:2016 certification in a design partner means documentation is embedded in the process - not retrofitted before submission.
✓ India-based partners with triple ISO certification can deliver regulatory-grade medtech prototyping at significantly lower cost than equivalent Western CDMOs.
The FDA classifies every medical device into one of three classes based on the level of risk it poses to patients.
Class I covers low-risk devices that do not support or sustain life. Think examination gloves, elastic bandages, and non-powered surgical instruments. These are subject to general controls - labelling, registration, and basic quality system requirements. Most are exempt from 510(k) premarket notification.
Class II covers moderate-risk devices that patients may depend on but that do not sustain life directly. Blood pressure monitors, infusion pumps, diagnostic imaging equipment, and most connected health devices fall here. Class II devices require general controls plus special controls, and most require a 510(k) premarket notification to the FDA before market entry.
Class III covers the highest-risk devices - those that support or sustain life, are permanently implanted, or pose an unreasonable risk of illness or injury. Pacemakers, cochlear implants, ventricular assist devices. Class III requires Premarket Approval (PMA) - the most rigorous pathway, requiring clinical trial data demonstrating both safety and effectiveness.
For EU and UK readers: the MDR 2017/745 framework uses a parallel system - Class I, IIa, IIb, and III - with CE Mark pathways that increase in rigor by risk level.
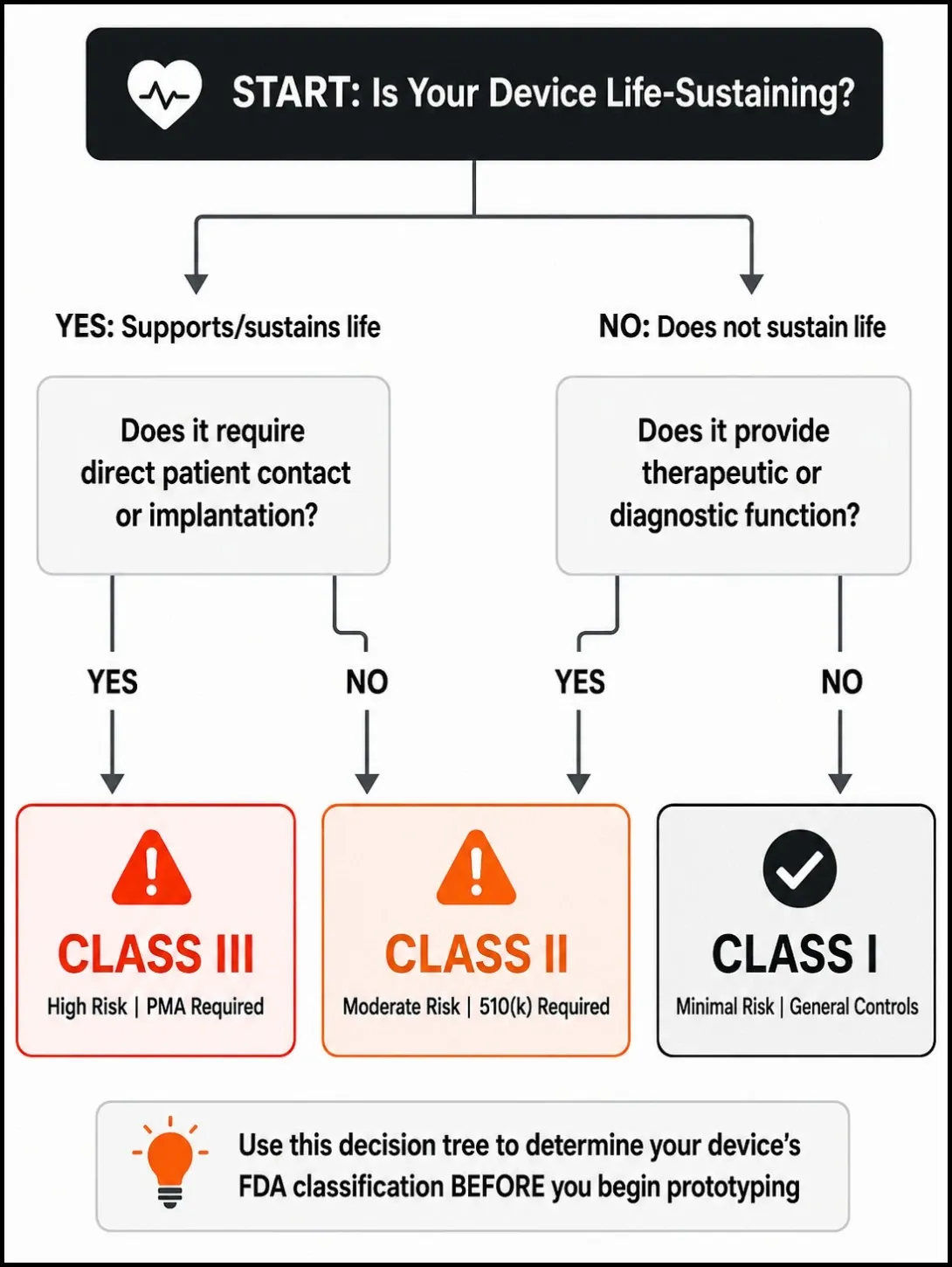
Under the FDA's Quality Management System Regulation (QMSR) and ISO 13485, design controls begin at the concept stage - not at the submission stage. That means the documentation obligation starts the moment you formalize your design inputs.
Your device's class determines:
✓ Which materials you can prototype in at each stage
✓ Which tests your prototype must pass
✓ What documentation you must generate during the build process
✓ How many prototype iterations regulators expect to see
✓ Whether your prototype can enter clinical or preclinical use
Choosing the wrong class or ignoring classification entirely does not just create paperwork problems. It creates engineering problems. You may prototype in materials that are not acceptable for your regulatory pathway, run tests that generate unusable data, or build a design history that does not match the submission format.
At iMAC, our 7-stage process starts at Design Research before any CAD work begins. Classification is confirmed at that stage, not discovered later.
Class I covers devices with minimal patient risk and no life-supporting function. Common examples include non-powered surgical instruments, medical gloves, hospital furniture, and certain diagnostic tools used externally.
"510(k) exempt" does not mean unregulated. Class I devices still require registration with the FDA, must comply with labeling requirements under 21 CFR Part 801, and must be produced under a quality system that meets general controls. What you are exempt from is the premarket notification - not compliance itself.
Class I gives you the most flexibility in prototyping. You do not need to use final production materials in early-stage builds. Representative materials that match the mechanical and physical properties of the intended final material are acceptable for alpha and beta stages.
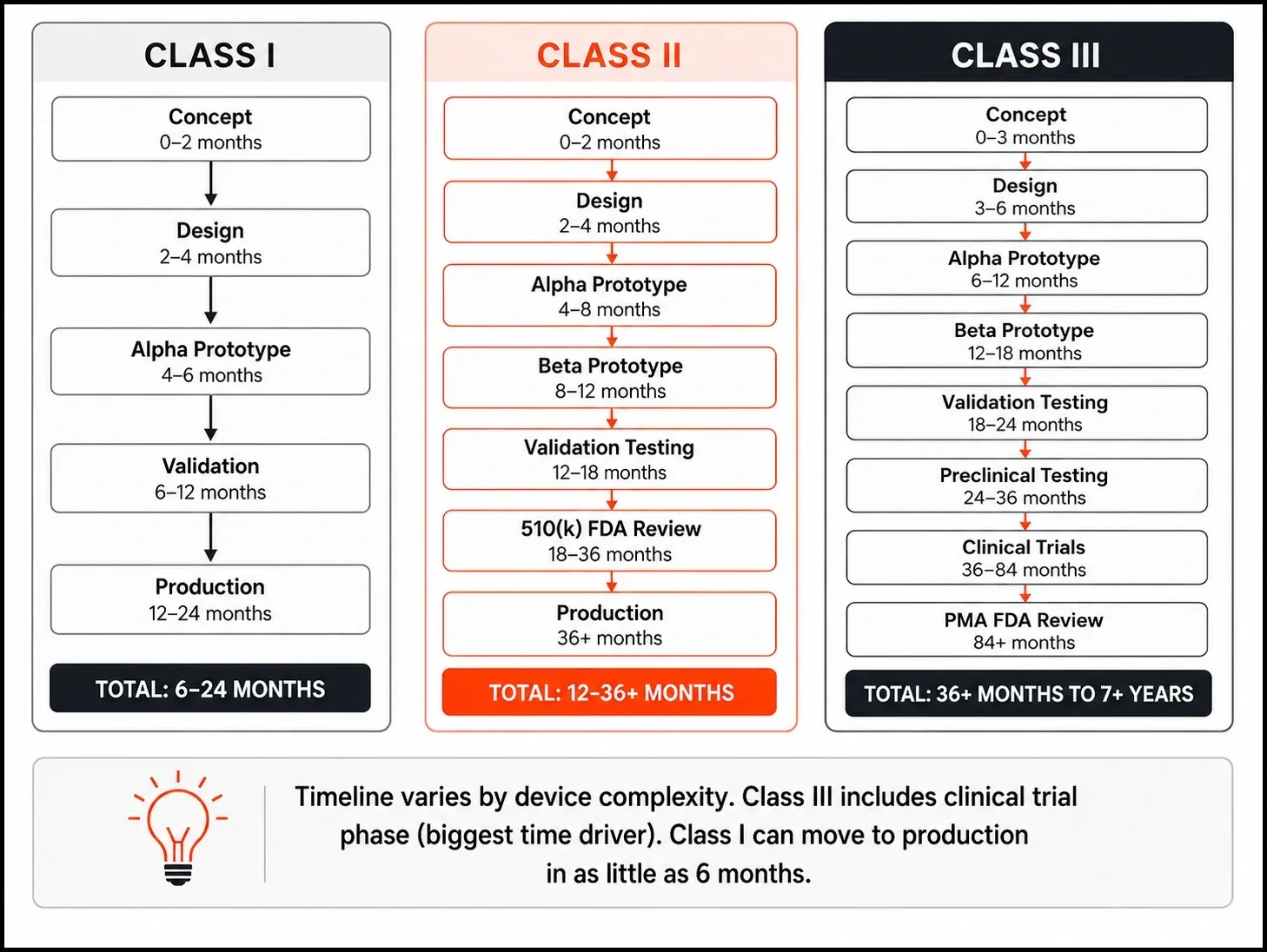
This means you can move fast. SLA 3D printing, FDM concept models, and vacuum-cast urethane parts - all are valid for early Class I prototyping. A well-scoped Class I device can move from concept to design freeze in 2 to 6 months.
Your prototype needs to prove three things: that it performs its intended function, that it works ergonomically for the intended user, and that it can be manufactured consistently.
Also Read: Top 6 Ways Rapid Prototyping Saves Development Time and Cost
The biggest mistake is treating "Class I" as "no compliance required." We see this regularly. Teams skip labeling reviews, build no design history, and produce no formal design outputs - then spend three to four months reconstructing documentation before they can register the device.
The second mistake is ignoring sterilization compatibility. If your Class I device will be supplied sterile, you need to validate the sterilization process against your prototype materials early - not after tooling is cut.
Build your DHF from prototype #1, even if you are not required to. It costs nothing to start and saves months before manufacturing.
Need faster Class I medical device prototyping without compliance gaps?
A 510(k) submission does not prove your device is better than what is on the market. It proves your device is substantially equivalent to a legally marketed predicate device - same intended use, same or different technological characteristics, with no new safety questions raised.
In prototyping terms, this means your prototype must generate test data that directly supports your substantial equivalence claims. Every performance test you run must be designed to answer the question: "Does this device perform at least as safely and effectively as the predicate?"
The FDA targets a 90-day review for 90% of 510(k) submissions. In practice, your timeline from prototype to 510(k) clearance runs 6 to 18 months - driven largely by how well your prototype documentation supports the submission.
Unlike Class I, Class II prototypes used in formal testing must be in near-final or final materials - especially for any component that contacts the patient. You cannot substitute prototype-grade resin for a patient-contacting polymer and submit biocompatibility data from the resin.
Your Class II validation prototype must generate:
✓ Functional performance data under simulated use conditions
✓ Biocompatibility evaluation under ISO 10993 (if any part contacts the patient)
✓ Electrical safety data to IEC 60601-1 (for active devices)
✓ EMC / electromagnetic compatibility data
✓ Human factors validation to IEC 62366-1
✓ Sterilization validation data (if applicable)
All of this must be documented in a Design History File.
Alpha vs. Beta vs. Validation Prototype: What Each Stage Must Achieve for Class II
Alpha prototype: Validates form and function. Materials can be representative. No regulatory testing yet. The goal is to confirm that the concept works and the design direction is correct. Think of it as your minimum viable product, the smallest build that tells you whether the core idea holds up before you commit to formal testing.
Beta prototype: Validates performance under simulated clinical conditions. Begin formal test protocols. Materials should be near-final for patient-contacting components. User testing and human factors data collection begin here.
Validation prototype: Final materials, final manufacturing process. This is the prototype that generates the formal V&V data submitted to the FDA. Design freeze applies from this point. At this stage, design for manufacturing decisions, tolerances, material selection, and assembly sequence must already be locked in. Discovering DFM issues after design freeze is one of the most common and costly delays in Class II development.
At iMAC, we have taken devices through this full progression. The Pill Cap medical IoT device moved from concept to injection molding production, with documentation generated at every stage. The Patient Warmer Igniva went through ICU validation. The documentation generated during prototyping became the foundation of the regulatory dossier.
What Triggers Class III Classification and Why PMA Is the Most Rigorous Pathway
Class III classification applies to devices that support or sustain human life, are permanently implanted, or present an unreasonable risk of illness or injury where general and special controls alone are insufficient. Pacemakers, cochlear implants, heart valves, and implantable neural devices are all Class III.
PMA is not a notification process. It requires you to prove with clinical evidence that your device is safe AND effective. The FDA has 180 days to review a PMA. In practice, the full development cycle runs 3 to 7 years, with total regulatory and testing costs ranging from $1M to $10M+.
How Class III Prototyping Is Fundamentally Different
For Class III, the prototype is not just a test build. It is a regulatory artifact.
Every design decision made during prototyping must be defensible in a PMA submission. Every material selection, every tolerance decision, every process parameter must be traceable from prototype to production. You cannot use a stand-in material for early prototypes and swap it out later without requalifying.
Before a Class III prototype can enter human clinical testing, you need an Investigational Device Exemption (IDE) from the FDA under 21 CFR Part 812. That IDE application itself requires bench testing data from your prototype. The prototype gates the clinical trial, not the other way around.
Bench Testing vs. Animal Studies vs. Clinical Trials: Understanding the Prototype's Role in Each
Bench testing is the first gate. Your prototype must demonstrate functional performance, mechanical integrity, and material properties under simulated worst-case conditions. A prototype that fails bench testing cannot advance.
Animal studies / preclinical testing require a prototype in final or near-final materials and production-equivalent processes. Biocompatibility, in vivo performance, and safety margins are evaluated here.
Clinical trials (IDE stage) require a clinical-grade prototype manufactured to the same specification as the intended production device. Any design change after the IDE is granted requires an IDE supplement and may trigger additional testing.
Choosing the Right Manufacturing Technology for Class III Prototypes
CNC machining in final production materials is the baseline for Class III implant prototypes. It produces verified material properties, tight tolerances, and no porosity risk. Metal additive manufacturing (DMLS/SLM) is increasingly used for complex implant geometries, but requires rigorous post-process characterisation and is not a substitute for verified material properties at the regulatory stage.
FDM 3D printing has no role in patient-contacting Class III prototype components.
| Dimension | Class I | Class II | Class III |
| Regulatory pathway | General controls / Exempt | 510(k) | PMA |
| Prototype materials | Representative acceptable | Near-final for V&V stage | Final production materials |
| Primary testing | Functional, ergonomic | Biocompatibility, EMC, usability | Full clinical-grade + IDE |
| DHF requirement | Recommended | Mandatory | Mandatory + full traceability |
| Typical timeline | 6 months – 2 years | 1 – 3 years | 3 – 7+ years |
| Prototype iterations (typical) | 2-4 | 4-8 | 6-15+ |
| Approx. development cost | $50K-$300K | $200K-$1M+ | $1M-$10M+ |
Cost ranges are industry-wide estimates for full development programs. Actual prototyping costs vary significantly by device complexity.
Not sure which medical device class your prototype falls under?
1. 3D Printing / Additive Manufacturing- Fastest for Early-Stage Validation
SLA and DLP printing produce fine surface detail and tight feature geometry - suitable for Class I and early Class II alpha prototypes. Resins require ISO 10993 biocompatibility characterisation before any patient-contacting use.
FDM is for concept models and ergonomic shells only. It has no place in regulatory testing for any class.
Metal DMLS/SLM is Class III-compatible with appropriate post-process characterisation and material qualification.
2. CNC Machining - Gold Standard for Validation Prototypes
CNC and VMC machining produce final-material, tight-tolerance parts suitable for validation testing across all three classes. For any component where mechanical properties must be verified, CNC machining is the standard. iMAC runs CNC and VMC machining in-house.
3. Vacuum Casting Cost - Effective Bridge for Class I and II
Vacuum casting uses silicone moulds to produce polyurethane parts that closely match injection-molded surfaces. Excellent for producing 5 to 50 units of near-final-quality plastic prototypes for ergonomic validation and limited user testing. Not suitable for patient-contacting use without a material qualification.
Typical lead time: 5 to 15 working days from CAD.
4. Injection Molding (Soft Tooling / Rapid Tooling)
Aluminum soft tooling produces actual injection-molded parts in production-intent materials. Used at beta and validation stages for Class II, and often required for Class III enclosure components. iMAC developed tooling and produced the Pill Cap through injection moulding in-house.
5. Sheet Metal Fabrication
Used for enclosures, brackets, and structural components in Class II capital equipment and Class III device housings. Combined with CNC machining for precision fits and assembly interfaces. iMAC fabricates sheet metal enclosures for medical device applications from its Ahmedabad facility.
Design History File (DHF) Your Prototype's Paper Trail
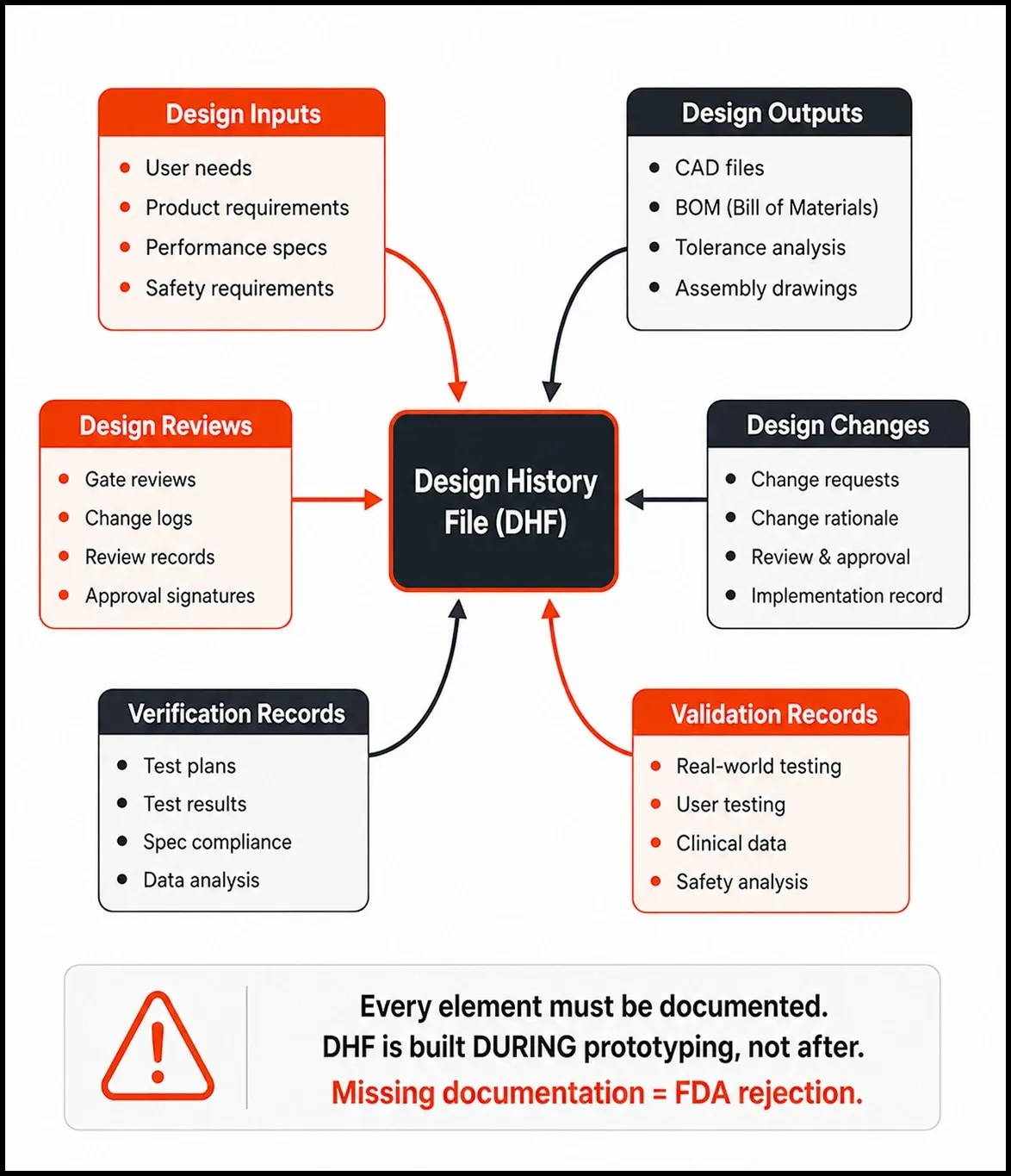
The DHF is required under 21 CFR Part 820 (harmonized as QMSR incorporating ISO 13485:2016 as of February 2026) and ISO 13485 Section 7.3. It must contain design inputs, design outputs, design reviews, verification records, validation records, and all design changes with rationale.
The most common DHF failure: engineers iterate the design without logging why the change was made. This breaks the traceability chain that FDA reviewers and auditors follow. Build the DHF during prototyping - not before the submission deadline.
Risk Management Under ISO 14971 - Starts at Prototype Stage
ISO 14971 risk management is not a document you produce at the end of development. It is an active engineering process. Risk analysis must begin at the design stage, and risk control measures must be validated on the prototype. Failure Mode and Effects Analysis (FMEA) should be updated with every iteration based on what testing reveals.
Verification & Validation (V&V) Testing on Prototypes
Verification answers: "Did we build it to spec?" It tests the prototype against design input specifications.
Validation answers: "Did we build the right thing?" It tests the prototype under real-world use conditions against user needs.
Both are mandatory under the QMSR and ISO 13485. Both must be documented in your Design History File. Biocompatibility evaluation (ISO 10993-1) and usability testing (IEC 62366-1) are both forms of validation performed on the prototype.
How iMAC's ISO 13485:2016 Process Embeds Documentation into Prototyping
iMAC Engineering holds ISO 13485:2016 certification for medical device quality management, alongside ISO 9001:2015 and ISO 27001:2013. This triple certification - rare for an Indian engineering firm of any size means that DHF-compatible documentation is produced during the prototyping phase, not retrofitted before submission.
Our 8-stage process – Design Research, Innovation & IP Strategy, Product Design, Engineering, Prototyping, Testing, Tooling, Manufacturing – generates a structured documentation trail at each gate. The NOX analgesia series moved from concept through manufacturing, with full documentation maintained at every stage. That documentation became the regulatory foundation - not an afterthought.
Class I Prototyping Cost Breakdown
✓ Concept to alpha prototype: $15,000 – $80,000, depending on complexity and electronics integration
✓ Validation prototype in final materials (injection moulded): $30,000 – $150,000
✓ Key cost drivers: enclosure complexity, electronics, tooling requirements
Class II Prototyping Cost Breakdown
✓ Alpha through validation prototypes, including formal testing: $80,000 – $400,000+
✓ 510(k) submission documentation and regulatory testing (EMC, biocompatibility, electrical safety): additional $50,000 – $200,000
✓ Testing lab costs alone: $30,000 – $100,000, depending on device type
Class III Prototyping Cost Breakdown
✓ Preclinical prototype development through IDE-ready stage: $200,000 – $1M+
✓ Full PMA development program across the full clinical cycle: $1M – $10M+
Working with an India-based ISO 13485-certified partner significantly reduces the engineering and prototyping spend at each stage without compromising the regulatory quality of outputs.
iMAC Engineering's Ahmedabad-based team delivers ISO 13485-aligned prototyping at a fraction of the cost of equivalent Western contract development organizations with USA and Canada virtual offices bridging the communication gap.
Looking for an ISO 13485-aligned prototyping partner with cost-efficient execution?
What "India Quality" Actually Means for Regulated Medical Devices in 2026
The assumption that India-based engineering means lower quality breaks down immediately when you look at certifications. ISO 13485:2016 certification requires a Quality Management System that meets the same international standard, whether the facility is in Ahmedabad, Boston, or Munich.
iMAC Engineering holds triple ISO certification: 9001:2015 (quality management), 13485:2016 (medical device quality management), and 27001:2013 (information security). That combination is rare globally, not just in India. It means your design IP is protected, your documentation meets international standards, and your regulatory dossier is built to a verified QMS.
What to Verify Before Choosing an Indian Medical Device Prototyping Partner
Before you sign an NDA with any Indian prototyping partner, ask for:
✓ ISO 13485:2016 certificate, not just ISO 9001
✓ Named medtech portfolio with device classes completed
✓ A sample design review record showing how documentation is produced during prototyping
✓ Embedded electronics and firmware capability, not just mechanical CAD
✓ IP protection process and ISO 27001 certification
Also Read: How to Choose the Best Product Design and Development Company?
Why iMAC Engineering is Built Specifically for Medtech Prototyping
iMAC Engineering has delivered 140+ projects across 30+ global clients in 5 years. Medical and healthcare is our lead vertical with named clients including Zydus, OmniBRx Biotechnologies, GITA Mediquip, Biotech, and Siemens Energy.
Our medtech portfolio spans ICU equipment, medical gas machines, surgical foot controllers, hospital infrastructure, and IoT-connected health devices. Every project moves through our 8-stage process with ISO 13485-aligned documentation at each stage.
We support clients through FDA and CE compliance pathways with an ISO 13485:2016-certified design process. We do not claim FDA facility approval, but we do build prototype programs that give your regulatory consultant a complete, well-structured DHF to work from.
Your device's class is not just a regulatory label. It is the engineering specification for your entire prototype program.
Get it right early, and every prototype iteration builds toward your regulatory submission. Get it wrong, and you spend 12 months rebuilding documentation that should have existed from day one.
The practical takeaway is straightforward: determine your device class before you build prototype #1, choose a design partner whose documentation process is aligned with your regulatory pathway, and treat the DHF as a live engineering document - not a compliance task.
iMAC Design & Engineering Services builds medical devices from concept through manufacturing - with ISO 13485:2016 certification, a proven portfolio across Class I, II, and Class III-adjacent devices, and an 8-stage process designed to generate regulatory-ready outputs at every stage.
If you are building a medical device and want to talk through your prototype strategy, reach out to our team.
Four to eight iterations are typical for Class II devices moving from proof-of-concept through alpha, beta, and validation. The exact number depends on design complexity, first-iteration quality, and the outcomes of user testing cycles.
It depends on the test and the component. SLA or metal DMLS prints with properly characterized material properties can be acceptable for some tests. For patient-contacting components, biocompatibility testing must be performed on the actual prototype material under ISO 10993. FDM prints are generally not acceptable for any regulatory testing. Confirm your approach with your regulatory consultant before committing to a prototype build strategy.
ISO 13485 does not accelerate FDA review timelines directly. But it significantly reduces the risk of documentation gaps that trigger FDA requests for additional information, which are the primary cause of 510(k) review delays. A partner operating under ISO 13485 generates documentation in a format that is already aligned with the FDA's QMSR requirements.
Verification confirms the prototype meets its design input specifications "Did we build it right?" Validation confirms the device performs safely and effectively for its intended use under real-world conditions "Did we build the right thing?" Both are required under QMSR and ISO 13485 Section 7.3, and both must be documented in your Design History File.
Immediately from your first formal design input document. The DHF is a live record of all design and development decisions, not a pre-submission document. Starting it at the prototype stage means you are building regulatory evidence in real time. Starting it just before submission means you are reconstructing decisions from memory, a process that creates credibility risk with FDA reviewers.
CNC machining in final production materials is the baseline for Class III implant prototypes. Metal additive manufacturing (DMLS/SLM) is increasingly used for complex geometries but requires extensive post-process characterization before it generates regulatory-acceptable data. All Class III prototypes used in preclinical or clinical testing must be manufactured using processes that are validated and transferable to production.
The FDA classifies devices based on risk level: Class I devices pose minimal risk (bandages, surgical instruments). Class II devices pose a moderate risk and typically require FDA clearance via 510(k) (infusion pumps, ECG monitors). Class III devices pose the highest risk; they support or sustain life, are permanently implanted, or present an unreasonable risk of harm and require Premarket Approval (PMA) via clinical trials (pacemakers, cochlear implants).
Timeline depends on your device's class. Class I typically takes 6-24 months from concept to production. Class II takes 12-36 months, with 6-18 months just for FDA 510(k) clearance. Class III requires 36+ months, with 3-7 years for the full PMA clinical trial pathway. Design and prototyping typically represent the first 4-8 months for any class, but regulatory and testing phases are what drive the longer timelines.
